phénomènes macroscopiques produits par les molécules d’eau
Ce paragraphe donne d’une façon qualitative et très simplifiée une explication à l’échelle moléculaire de phénomènes globaux (pression osmotique d’une solution, effets capillaires...) qui seront examinés dans les chapitres suivants d’une façon plus théorique. Le lecteur pourra s’appuyer sur cette présentation pour garder une vision concrète des lois physiques présentées.
Un ensemble de dipôles s’organise spontanément en un réseau lâche.
Les forces d’attraction (entre charges contraires) et de répulsion (entre charges semblables) amènent les molécules, dans l’eau liquide, à se disposer statistiquement de telle sorte que, à chaque instant, les premières soient maximisées et les secondes minimisées. Néanmoins les forces d’attraction intermoléculaires restent faibles.
Au sein de la masse liquide, les forces d’attraction exercées sur une molécule par toutes celles qui l’entourent se compensent par symétrie (fig. 6a). En revanche, les molécules situées à la surface du liquide ne subissent des forces d’attraction que dans le plan de la surface et en dessous : il en résulte une attraction non uniforme des molécules dans la masse du liquide et à sa surface (fig. 6b). Ces molécules en surface sont plus fortement liées entre elles que les molécules situées plus en profondeur et constituent une sorte de « peau » superficielle, visible sur une goutte d’eau. Cette force de surface qui porte le nom de « tension superficielle » s’exerce dans tous les phénomènes liés à la capillarité (chap. I.3), c’est-à-dire à l’ensemble des phénomènes qui se produisent à la surface des liquides (dans les capillaires notamment). Dans la glace, la structure variable du réseau des dipôles apparaît dans la géométrie des flocons de neige par exemple (réseau à base hexagonale) (fig. 4c).
Remarque : la molécule d’eau étant un dipôle est elle-même attirée par les molécules polaires. Quand l’autre molécule polaire est une molécule d’eau on parle de cohésion. Cette cohésion est à l’origine de la tension de surface de l’eau dont on a parlé ci-dessus. Quand au contraire il s’agit d’une molécule d’une espèce différente, on parle d’adhésion.
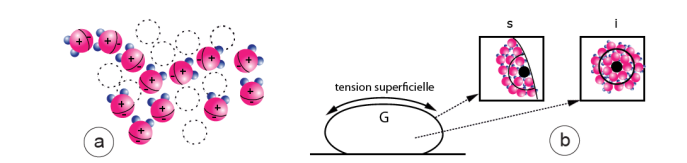
Figure 6. a) Au sein de l’eau liquide, les molécules dipôles sont statistiquement disposées les unes par rapport aux autres de telle sorte que leurs deux pôles sont plus proches des pôles opposés de leurs voisines (attraction) que des pôles de mêmes charges (répulsion).
b) Dans une goutte d’eau (G) une molécule de surface (s, en noir), est soumise à des forces issues d’une demi sphère, tandis qu’une molécule de l’intérieur (i, en noir) est soumise à des forces issues d’une sphère concentrique entière. Cette dissymétrie des forces en surface et en profondeur est à l’origine des phénomènes de tension superficielle et de capillarité.
L’agitation thermique bouscule en permanence les réseaux de dipôles dans l’eau liquide.
Quelle que soit la température[10] à laquelle est soumise un corps, ses molécules (et les atomes qui forment ces molécules) s’agitent continûment, d’autant plus que la température augmente. Dans un gaz ou un liquide l’agitation est désordonnée (fig. 7a). Dans un solide, elle se fait autour d’une position d’équilibre. C’est ce qu’on appelle l’agitation thermique, mise en évidence d’une façon simple mais spectaculaire par le mouvement brownien[11]. Cette agitation thermique s’accroît en amplitude avec la température. C’est elle qui, à température ambiante, permet à un flux de molécules d’eau de s’échapper en permanence de la surface liquide, dans l’évaporation (fig. 7b) ; c’est elle qui, à la température dite d’ébullition, fournit à ces molécules assez d’énergie d’agitation pour s’échapper, sous forme de vapeur, dans tous les points de la masse liquide (fig. 7c). A toute température, l’agitation thermique produit des chocs continuels entre les molécules. Ces chocs provoquent la rupture de certaines liaisons qui se rétablissent aussitôt après avec d’autres dans un mouvement incessant.
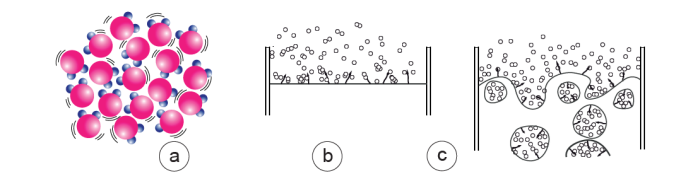
Figure 7. L’agitation thermique entre dans l’explication de plusieurs phénomènes affectant l’eau liquide.
a) Schéma suggérant l’agitation thermique (modérée) au sein de l’eau liquide ; cette agitation augmente avec la température. Elle a tendance à séparer les molécules.
b) L’évaporation se produit à la surface de l’eau liquide parce que l’énergie des chocs dus à l’agitation thermique peut dépasser celle qui lie entre elles les molécules dipôles.
c) À la température de l’ébullition, le passage de l’eau liquide à l’eau vapeur se produit, cette fois, dans toute la masse du liquide
(cf. bulles de vapeur dans le liquide).
[10] Sauf au zéro absolu.
[11] R. Brown (1795-1858). Ecossais, médecin de formation, il participe à un voyage d’exploration le long des côtes australiennes de cinq ans ; il en revient avec plus de 4000 espèces de plantes et se voit confier quelques années plus tard le poste de conservateur au British Muséum. L’un des premiers à utiliser couramment le microscope, il entreprend des études de classification botanique par la forme des grains de pollen. Il observe alors la présence de très petites particules issues de ce pollen, bougeant dans tous les sens. Croyant dans un premier temps qu’il s’agit d’une propriété d’un « fluide vital » il renouvelle l’opération avec des particules anorganiques et change d’avis attribuant ces mouvements aux particules elles-mêmes. Il publie ses observations en 1828 en précisant que l’explication de ces mouvements est physique et non pas biologique, ce qui était une révolution à son époque. L’explication précise de ces mouvements ne sera donnée que bien plus tard : si on place de très fines particules (mais bien plus grosses que des molécules) à la surface d’un fluide, elles sont soumises à un bombardement incessant par les molécules qui constituent le fluide. Ces chocs incessants venant de toutes les directions se traduisent par un mouvement aléatoire des particules, appelé mouvement brownien. Bien plus tard (au début du 20ème siècle), J. Perrin, A. Einstein et M. Smoluchowski parmi d’autres, feront la théorie de ce mouvement brownien qui du coup, deviendra l’une des meilleurs preuves de la réalité moléculaire (Duclaux, 1938)
Les molécules d’eau établissent des liaisons avec les solutés.
Les substances solides solubles dans l’eau (ex : glucose, chlorure de sodium) subissent au sein de ce liquide des chocs incessants de la part des molécules d’eau. Chaque molécule arrachée à la surface solide est aussitôt entourée d’une « coque » de molécules d’eau avec lesquelles s’établissent des liaisons de forces très variables. Si le soluté s’ionise[12] en se dissolvant, les liaisons fortes dipôles anions et dipôles cations[13] maximisent les forces d’attraction (fig. 8). Si le soluté offre lui aussi une structure de dipôle en raison de la disposition électronique de surface, c’est encore la maximisation des forces d’attraction mutuelle qui organisera les dipôles H2O. Si enfin, le soluté ne présente aucune charge électrique, mais seulement des radicaux[14] ayant des affinités pour l’eau (ex : radicaux hydroxyles 0H) les liaisons auront une intensité plus faible encore.
Une forte liaison eau-soluté (plus forte que la liaison eau-eau) diminue notablement la liberté de mouvement des dipôles H2O, directement au contact du soluté, mais agit au-delà de cette première couche sur les couches concentrées de dipôles, avec une intensité qui décroît avec la distance (fig. 9). Cette perte de liberté est assimilée à une perte d’énergie de l’eau et à une diminution de ce qu’on appelle son « potentiel chimique » (cf. chap. I.2). Un soluté abaisse d’autant plus le potentiel chimique d’une solution que la concentration de celle-ci est élevée.
Lorsqu’une substance (soluté) se dissout dans un liquide (solvant) au repos (fig. 10), c’est l’agitation thermique qui assure seule la dispersion progressive des molécules de soluté dans toute la masse du liquide : la solution s’homogénéise par diffusion. Celle-ci est d’autant plus lente que la température est basse. Lorsque l’homogénéisation est assurée par un mouvement du liquide, le phénomène assurant le mélange porte le nom de convection.
Mettons en relation deux compartiments, l’un contenant de l’eau pure et l’autre un soluté en solution, en les séparant par une membrane imperméable au soluté mais pas à l’eau. On constate que l’eau se déplace du compartiment d’eau pure vers celui contenant les solutés. Ce mouvement de l’eau s’appelle l’osmose. Il est étudié en détail au chapitre I.2.
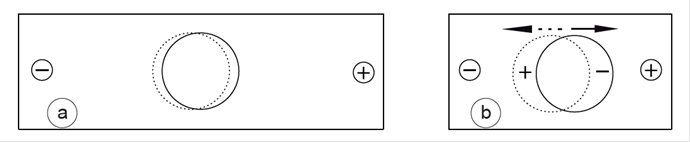
Figure 8. a) La molécule-dipôle s’oriente par rapport aux charges électriques (+ et -) qui l’environnent.
b) Lorsqu’elle s’en rapproche, sa coque électronique se déforme, le défaut de recouvrement des champs électriques du dipôle (c’est-à-dire le fait qu’ils ne soient pas exactement superposés) augmente en même temps que ses propres charges.
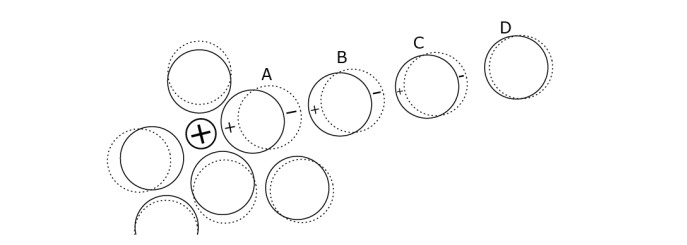
Figure 9. Les molécules d’eau sont fortement attirées par les solutés ionisés. Elles forment autour d’eux des couches concentriques. La charge des dipôles est d’autant plus élevée que ceux-ci sont plus proches de l’ion (ici un cation, marqué d’une croix) comme le dipôle A. La charge des dipôles B, C, D, décroît avec l’éloignement, jusqu’à la valeur qu’elle a dans la masse d’eau pure.

Figure 10. L’eau et les solutés. a) Du sucre placé au fond d’un verre d’eau pure se dissout d’abord en une solution concentrée, dont les molécules vont progressivement diffuser dans tout le liquide jusqu’à ce que la solution soit homogène. La diffusion est un phénomène lent dû à l’agitation thermique. Sa vitesse s’accroît avec la température.
b) Un train de bulles d’air montant dans un aquarium crée un mouvement de convection de l’eau qui maintient l’oxygène dissous à une concentration constante.
c) Une cloison poreuse à l’eau mais imperméable à des molécules de solutés plus volumineuses, permet à l’eau de pénétrer dans le compartiment de droite, dont le niveau augmente avec le temps. C’est le phénomène d’osmose. Ce passage de l’eau dure jusqu’à ce que la différence de niveau compense la pression osmotique. Voir chap I.2 pour une présentation détaillée de l'osmose.
[12] C’est-à-dire qu’il perd sa neutralité électrique par acquisition ou perte d’un ou plusieurs électrons.
[13] Un anion est un ion à charge négative, un cation est un ion à charge positive.
[14] On appelle radicaux de petites unités d’atomes différents constitutives des molécules. L’éthanol (C2H5OH) par exemple, rassemble les radicaux CH3, CH2, OH.
Les molécules d’eau établissent des liaisons avec les surfaces solides non solubles.
La majorité des constituants minéraux et organiques des sols sont quasiment insolubles dans l’eau, de même que les tissus végétaux. Ces constituants et ces tissus offrent au contact de l’eau des surfaces très étendues par unité de masse : on cite fréquemment le cas des argiles dont quelques grammes développent une surface égale à celle d’un terrain de football. De plus ces surfaces ont une affinité pour l’eau (une hydrophilie) en ce sens que les liaisons eau-surface sont plus fortes que les liaisons eau-eau des molécules. Plusieurs raisons à cela : les surfaces minérales, issues de brisures de réseaux ioniques cristallins, présentent des charges positives et négatives. Les champs de force qui leur sont associés sont imparfaitement recouverts (même situation que ce qui se passe dans la molécule d’eau) si bien qu’elles se présentent comme des « multipôles » ; de ce fait, elles attirent les dipôles H2O, ici par leur extrémité positive, là par leur extrémité négative.
Pour leur part, les surfaces des constituants organiques, ceux du sol (issus principalement de la décomposition des tissus végétaux) et ceux des tissus vivants des plantes, présentent en abondance des fonctions chimiques ionisables[15] (ex : fonctions carboxyliques[16]) ou des fonctions aptes à l’établissement de liaisons hydrogène avec l’eau (fonctions hydroxyles[17]). Des molécules ainsi retenues sur une surface sont dites adsorbées (fig.11b et 11c).
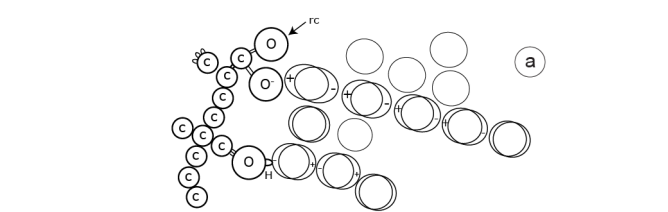
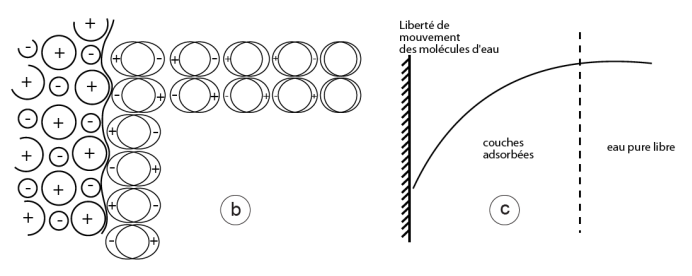
Figure 11. Phénomènes d’interface.
a) Représentation schématique d’une portion de surface formée d’un réseau d’ions d’un minéral insoluble. Les molécules dipôles d’eau s’adsorbent en couches plus ou moins régulières et sont d’autant plus liées qu’elles sont plus proches de la surface comme dans le cas de la figure 9.
b) Même type de représentation mais pour des surfaces organiques, lesquelles sont irrégulières et non rigides. Un radical carboxylique (-COO-) ionisé ainsi qu’un radical hydroxyle (-HO-) sont figurés ; rc : radical carboxylique.
c) La « liberté » de mouvements moléculaires de l’eau, maximale au sein de l’eau pure diminue d’autant plus que les molécules sont plus proches d’une molécule de soluté (fig. 9) ou de la surface, s, qui les attire. Ces molécules d’eau aux mouvements plus ou moins restreints forment des couches d’eau dites adsorbées. A l’échelle macroscopique cette plus ou moins grande liberté de mouvements se traduit en terme de potentiel chimique ou matriciel (voir chapitre I.2).
De plus, les constituants du sol et des tissus végétaux forment des structures poreuses. Les pores, innombrables et interconnectés, présentent des dimensions (rayons moyens) très variables, entre le micromètre et le millimètre. Par capillarité (chap. I.3) l’eau remplit ces pores. Etant donné que le rapport surface sur volume des pores est d’autant plus élevé que ceux-ci sont plus fins, il en résulte que l’énergie de rétention de l’eau s’accroît avec la finesse de ces pores. La rétention de l’eau par les surfaces poreuses hydrophiles diminue la liberté de mouvement de ses molécules. Cette rétention change l’état énergétique des molécules d’eau. Pour des raisons semblables, le même phénomène se produit avec les molécules d’eau d’une solution : leur état énergétique est affecté par la présence du soluté. Dans les deux cas, mais pour des raisons différentes, la présence de surfaces poreuses ou la présence de solutés, l’état énergétique des molécules d’eau a changé. A l’échelle macroscopique, où l’on ne tient plus compte des énergies de liaison différentes d’une molécule à l’autre mais d’un effet moyen sur des milliards et des milliards de molécules, on dira que le potentiel chimique de l’eau a varié (chap. I.2).
Sommaire :


